


В ИОХ РАН получены новые стабильные 2-фтораллильные электрофилы
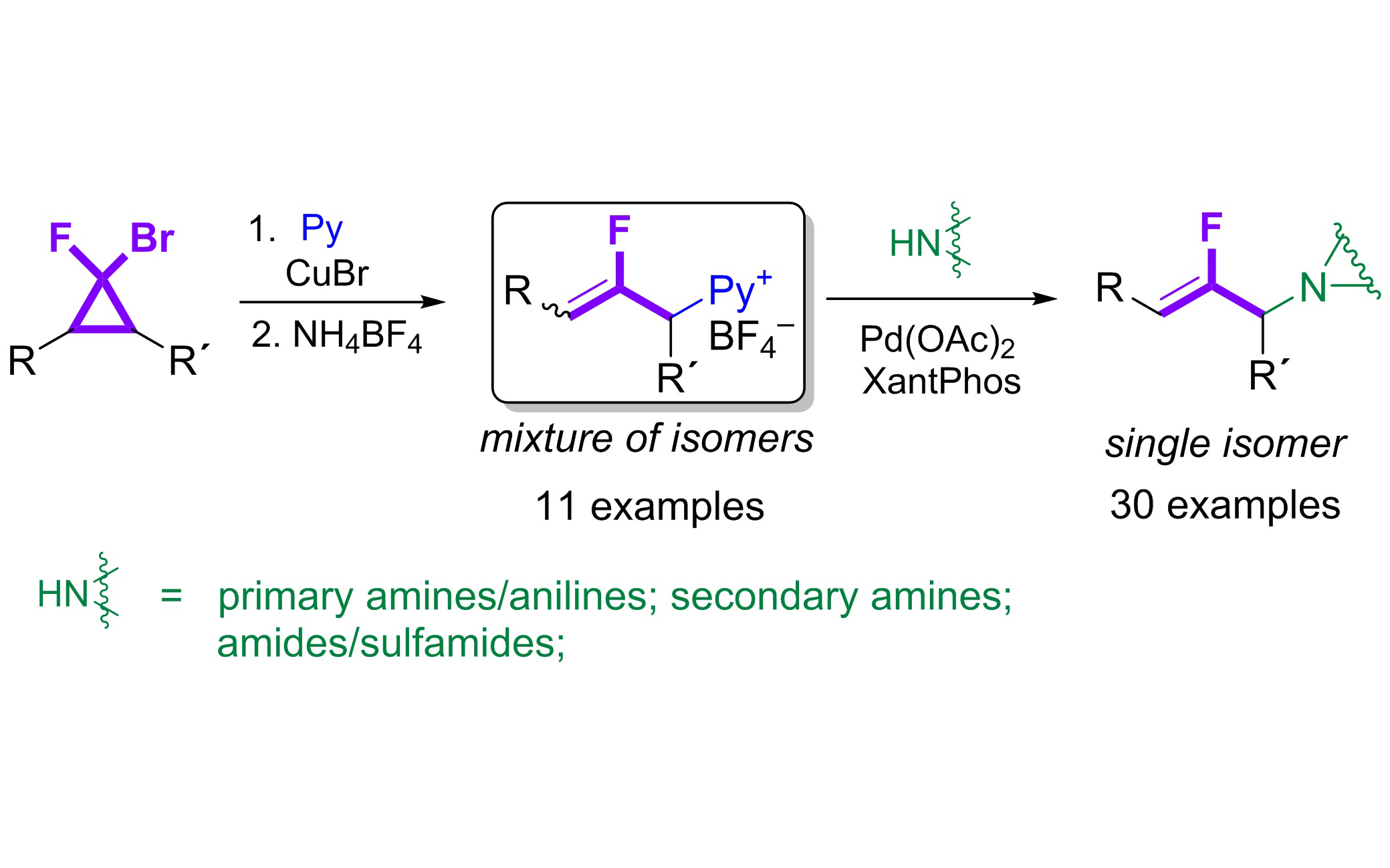
Фторированные биоизостеры нашли широкое применение в создании современных лекарственных препаратов. В частности, фторалкеновый фрагмент может эффективно имитировать амидную связь из-за сходства электронных и стерических параметров, тем самым увеличивая гидролитическую стабильность, липофильность и проницаемость молекул через мембраны. Используя такую биоизостерическую замену, можно существенно повысить эффективность известных на сегодняшний день биологически активных соединений.Среди методов получения фторалкенов и соединений, содержащих 2-фтораллильный фрагмент, одним из наиболее удобных является раскрытие цикла во фтор-замещенных циклопропанах. Однако его синтетическое применение долгое время ограничивалось по причине сложности контроля регио- и стереоселективности присоединения нуклеофила к 2-фтораллил-катионным структурам, которые являются ключевыми промежуточными продуктами в этих процессах.
В Лаборатории химии диазосоединений ИОХ РАН путем раскрытия гем-бромфторциклопропанов синтезированы тетрафторбораты (2-фтораллил)пиридиния — новые стабильные 2-фтораллильные электрофилы. Реакции этих солей с аминами, малонатами и бороновыми кислотами, катализируемые палладием, открывают путь к широкому ряду функционализированных соединений с фтораллильным фрагментом. Все обнаруженные превращения протекают с высокой регио- и стереоселективностью.
Источник:
Angelina Yu. Bobrova, Maxim A. Novikov, and Yury V. Tomilov (2-Fluoroallyl)pyridinium tetrafluoroborates: novel fluorinated electrophiles for Pd-catalyzed allylic substitution Org. Biomol. Chem., 2021, 19, 4678-4684. DOI: 10.1039/D1OB00567G.