Theoretical Chemistry Group (No. 24)

Telegram-канал: medvedev_m_g
For inquiries: @MMG_assistant on Telegram
ORCID: 0000-0001-7070-4052
ColabID: R-386BE-0850A-CG10N
Researcher ID: N-3097-2016
Google Scholar: Michael G. Medvedev
h-index: 21
- Modeling of Chemical Processes: Simulation of chemical reactions (OrgChemFront2025, JACS2025, TetrahedChem2024, JOC2024, JOC2023, OPRD2023, EJOC2022, JACS2022, JACS2021, JACS2020, ANIE2017), biological processes (JCIM2023, JACS2017), photochemical processes (ChemEur2023, D&P2022, D&P2019), electrochemical processes (ElectrochemActa2025, OrgChemFront2024), and solid-state processes (ACSApplMatInt2025, JPCLett2021).
- Development of Theoretical Chemistry Methods. Theoretical chemistry methods include all computational tools used in direction 1: density functional theory (DFT), conformational search algorithms, molecular modeling automation, force fields, basis sets, etc. Within this direction, the group tests existing approaches (JPCA2025, JPCLett2024, Science2022, ChemSocRev2021, Science2017), develops new ones (JCIM2025, JCP2025_1, JCP2025_2, JCIM2024, JCIM2023, MendComm2017), designs general frameworks for creating new methods (JPCA2024 (for DFT), WCMS2023 (for conformational search)) and refines software packages for specific applications (JCP2025_1, MendComm2021).
- Applications of Artificial Intelligence in Chemistry:AI is used to accelerate and refine molecular modeling (JCIM2025, Science2022), and to address other challenges in chemical research, such as retrosynthesis (our development within the IHR RAS Megagrant) and detector signal correction (ACSApplPolyMat2024).
- Modeling of Neural Interaction Processes: Although not directly related to chemistry, this research direction stems from our interest in understanding thought processes and neuromorphic computing. It includes two subtopics: developing neuromorphic networks to assist projects in directions 2 and 3, and exploring how biological neural networks learn and function. The group builds spiking neural network models to investigate emergent principles of learning and behavior.
✓ In 2024–2025, an AI-based method was developed to help identify “missing” molecular conformations — stable spatial structures that are often overlooked during modeling. The new approach combines quantum-chemical calculations with machine learning (Gaussian processes) to analyze molecular geometries generated by other conformational search methods and efficiently uncover the missing ones in just 20–30 iterations.
When tested on 60 potentially biologically active compounds, the algorithm discovered up to 28 previously undetected conformations (geometries) for nearly half of the molecules. This method significantly improves the accuracy of molecular modeling and accelerates the discovery of potential drugs and catalysts. JCIM2025

✓ In 2023–2024, a conformational search algorithm based on the inverse kinematics method, Ringo, was developed and implemented (https://github.com/TheorChemGroup/Ringo). The algorithm enables the analysis of conformational flexibility in any polycyclic molecule using inverse kinematics and identifies the molecule’s degrees of freedom — that is, the set of dihedral angles capable of independent rotation while keeping bond lengths and valence angles fixed.
The developed algorithm outperforms all alternative methods for conformational sampling of cyclic molecules — including metadynamics, distance geometry, and LowModeMD — in both speed and efficiency of exploring the configurational space of polycyclic systems. JCIM2024, WCMS2023

✓ In 2024, a new approach was developed to assess uncertainty in the product ratio of chemical reactions modeled using molecular dynamics. This method revealed errors in previous studies and will, in the future, help improve the reliability of both qualitative and quantitative conclusions in such simulations.JPCLett2024

✓ In 2023, in collaboration with Fyodor Novikov’s group, an algorithm was developed to calculate the relative biological activities of bioisosteric molecules with accuracy comparable to experimental data.
Bioisosteres are molecules that differ by substituents but share similar conformational profiles, allowing them to bind to proteins in the same way. Bioisosteric substitutions are typically performed to improve metabolic stability and increase compound activity. The developed method enables fast and accurate identification of which hydrogen-to-fluorine substitutions in a molecule enhance its biological activity. JCIM2023

✓ In 2023, in collaboration with colleagues from Laboratory No. 110 at INEOS RAS, an experimental and theoretical study was carried out on the photoisomerization of cymanthrene–quinazolinone derivatives. For the first time, it was demonstrated that the cage effect of the solvent can control the outcome of a photochemical reaction involving a cymanthrene derivative.
Quantum-chemical calculations revealed a reaction mechanism that involves photoinduced CO dissociation followed by rapid reassociation within the solvent cage. This mechanism was later confirmed experimentally: when the solution was irradiated with ultrasound, which disrupts solvent cages, the recombination of CO was suppressed, steering the reaction toward a specific product. ChemEur2023

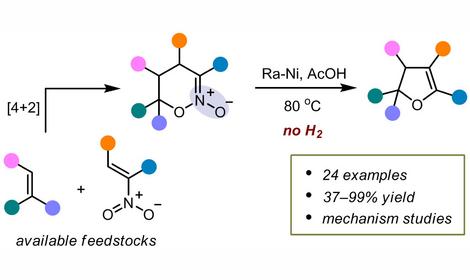
✓ In 2020-2023, members of the Group modeled a series of reactions—developed in Laboratory No. 13 at IOC RAS—for the synthesis of a new class of compounds: cage-like organic peroxides, which are promising for pharmaceutical and agrochemical applications.
Using quantum-chemical modeling, the mechanisms of these reactions were elucidated, and the main puzzle of this class of processes—their unusually high selectivity—was solved. It was found that this selectivity arises from the inverse α-effect.
This discovery makes the assembly of organic peroxides predictable and paves the way for faster and more efficient design of new cage-like molecules in the future. JOC2023, JACS2022, JACS2021, JACS2020

7) In collaboration with colleagues from Laboratory No. 13 at IOC RAS, Florida State University (USA), and Bu-Ali Sina University (Iran), a comprehensive comparison of the main models of stereoelectronic effects—orbital, electrostatic, and steric—was carried out.
It was shown that, although the total molecular energy arises from many contributions (bond and orbital interactions, electrostatic forces, and steric repulsion), the patterns of change in orbital interactions possess the greatest predictive power for describing chemical reactivity.
Furthermore, key stereoelectronic interactions were identified for the most common classes of organic compounds, and systematic trends in their influence on chemical and physical properties were established. Such oxygen-involving stereoelectronic effects can be harnessed by chemists to design new reactions and control their mechanistic pathways. ChemSocRev2021_1, ChemSocRev2021_2

8) In 2022, a commentary was published in Science demonstrating that a 2021 paper by the British artificial intelligence company DeepMind provided insufficiently reliable evidence for the correctness of its DM21 functional when applied to systems containing a non-integer number of electrons.
Reliable quantum-chemical predictions can replace some experimental work involving expensive reagents with simulations of digital analogs of real chemical systems. However, to ensure the accuracy of such predictions, density functionals must be capable of correctly handling fractional electron numbers. Science2022

9) In 2017, Michael Medvedev and co-authors developed a methodology for evaluating the quality of electron densities in atomic systems obtained from density functional theory (DFT) methods.
Using this approach, it was shown that the most popular DFT methods at the time were overfitted and could be unreliable when modeling systems or properties significantly different from those used in their training. The study also identified robust, non-overfitted DFT functionals, including PBE0, B3PW91, B98, TPSS, SCAN, OLYP, and others. Science2017